nf-core/circrna
circRNA quantification, differential expression analysis and miRNA target prediction of RNA-Seq data
Introduction
nf-core/circrna is a bioinformatics pipeline to analyse total RNA sequencing data obtained from organisms with a reference genome and annotation. It takes a samplesheet and FASTQ files as input, performs quality control (QC), trimming, back-splice junction (BSJ) detection, annotation, quantification and miRNA target prediction of circular RNAs.
The pipeline is still under development, but the BSJ detection and quantification steps are already implemented and functional. The following features are planned to be implemented soon:
- Isoform-level circRNA detection and quantification
- circRNA-miRNA interaction analysis using SPONGE and spongEffects
- Improved downstream analyses
If you want to contribute, feel free to create an issue or pull request on the GitHub repository or join the Slack channel.
Pipeline summary
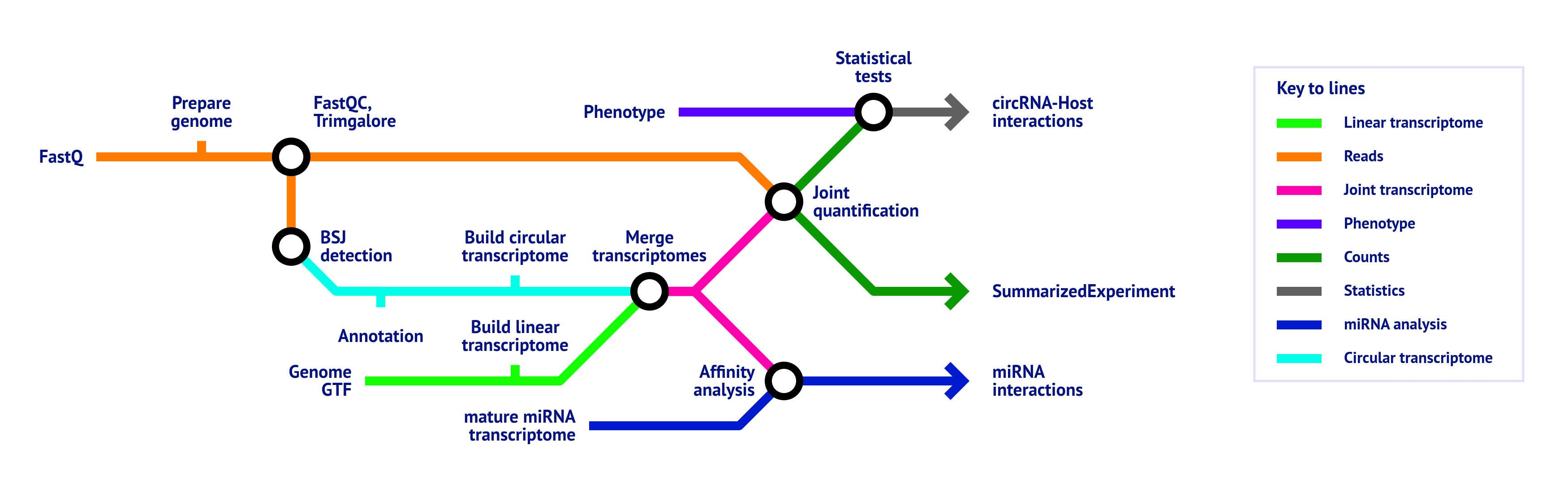
- Raw read QC (
FastQC) - Adapter trimming (
Trim Galore!) - BSJ detection
- circRNA annotation
- Based on a GTF file
- Based on database files (if provided)
- Extract circRNA sequences and build circular transcriptome
- Merge circular transcriptome with linear transcriptome derived from provided GTF
- Quantification of combined circular and linear transcriptome
- miRNA binding affinity analysis (only if the
matureparameter is provided)- Normalizes miRNA expression (only if the
mirna_expressionparameter is provided) - Binding site prediction
- Perform majority vote on binding sites
- Compute correlations between miRNA and transcript expression levels (only if the
mirna_expressionparameter is provided)
- Normalizes miRNA expression (only if the
- Statistical tests (only if the
phenotypeparameter is provided) - MultiQC report
MultiQC
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow.Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
sample,fastq_1,fastq_2
CONTROL,CONTROL_R1.fastq.gz,CONTROL_R2.fastq.gz
TREATMENT,TREATMENT_R1.fastq.gz,TREATMENT_R2.fastq.gzEach row represents a fastq file (single-end) or a pair of fastq files (paired end).
Now, you can run the pipeline using:
nextflow run nf-core/circrna \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters;
see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
nextflow run nf-core/circrna \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/circrna was originally written by Barry Digby. It was later refactored, extended and improved by Nico Trummer.
We thank the following people for their extensive assistance in the development of this pipeline (in alphabetical order):
Acknowledgements

Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #circrna channel (you can join with this invite).
Citations
nf-core/circrna: a portable workflow for the quantification, miRNA target prediction and differential expression analysis of circular RNAs.
Barry Digby, Stephen P. Finn, & Pilib Ó Broin
BMC Bioinformatics 24, 27 (2023) doi: 10.1186/s12859-022-05125-8
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.