The nf-core/callingcards workflow is here!

What is Calling Cards?
Calling cards is the name of a sequencing based assay developed in Rob Mitra’s lab (https://mitralab.wustl.edu/) at Washington University in St Louis to interrogate the DNA binding locations of proteins called Transcription Factors. Transcription Factors (TF) are a class of proteins capable of binding to DNA to affect the transcription of genes. In order to record where a given TF binds, calling cards harnesses the power of a retrotransposon, or a transcribed genomic sequence capable of copying itself back into the genome, to deposit a molecular barcode into the genome when a TF binds.
Calling Cards in Yeast
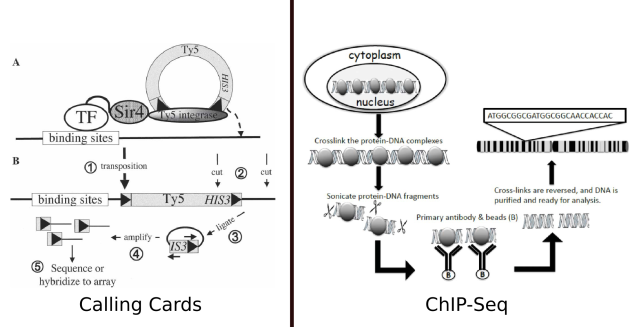
Figure 1: A comparison of Calling Cards to the more familiar method of interrogating transcription factors, ChIP-Seq. The calling cards figure is from Calling cards for DNA-binding-proteins by Wang et al., which originally described calling cards. The ChIP-seq diagram is from
Role of ChIP-seq in the discovery of transcription factor binding sites
by Mundade et al.
In the yeast version of calling cards, a retrotransposon complex is linked to the TF of interest. When the TF binds to the genome, the Ty5 sequence is inserted, leaving behind a “calling card” which records the presence of that transcription factor at that site. In contrast to calling cards, which provides a longitudinal record of TF/DNA interaction, in ChIP experiments, the protein are cross-linked to the DNA, in effect providing a snapshot of the state of the DNA and its associated protein at a single point in time.
Calling Cards in Mammals
Much of the current work and development on the Calling Cards assay has focused on the mammalian system. This differs in technical detail from the yeast procedure, though the outcome is similar. Figure 2 below provides a graphic overview of the biochemical procedure.
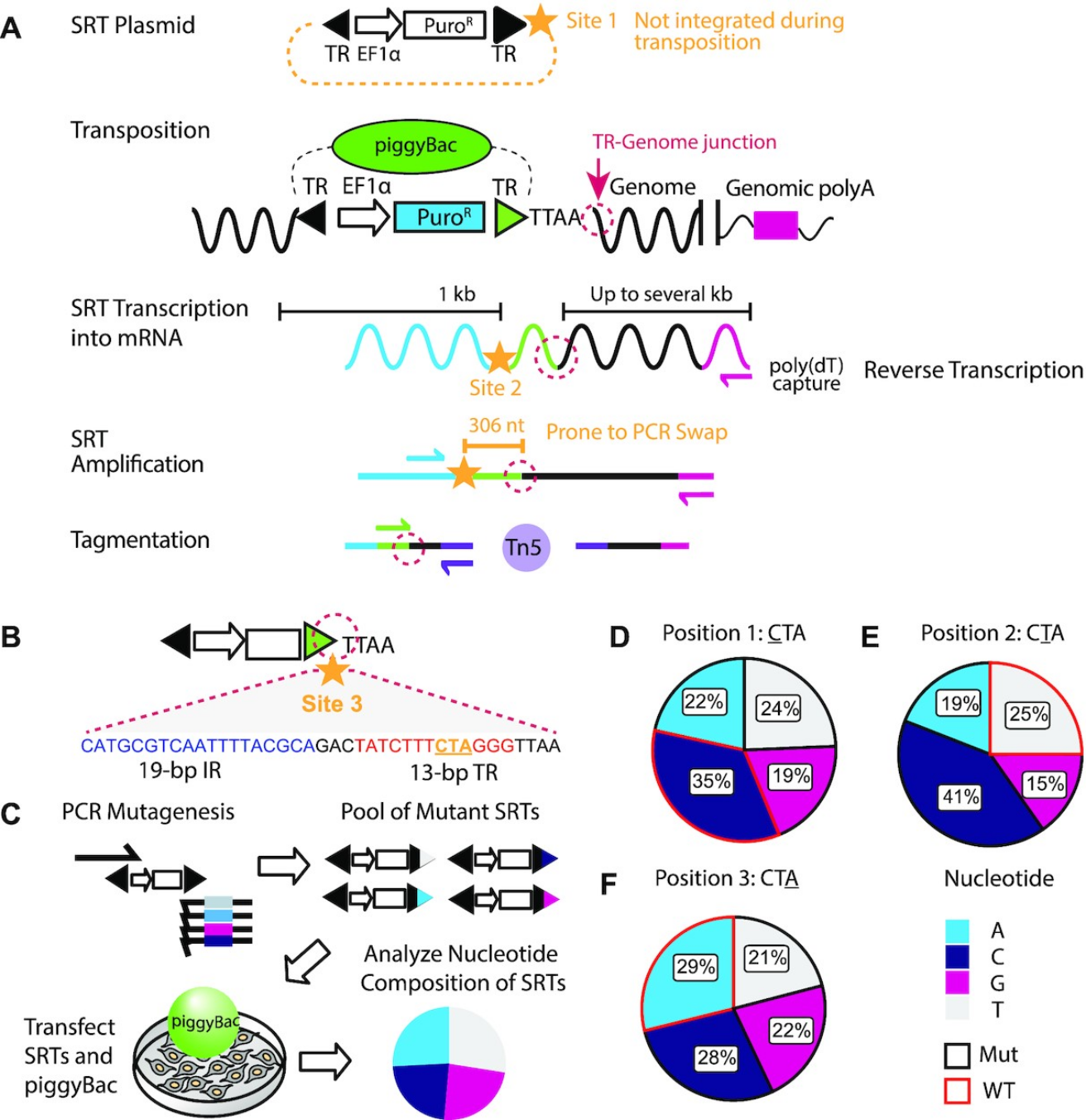
Figure 2: Barcoding the self-reporting transposon. (A) Schematic overview of the SRT construct, Calling Card
method, and sequencing library preparation. Candidate sites for barcode insertions are indicated with gold
stars. The TR-Genome junction, used to map transposon insertions, is circled in dotted magenta line. (B) Barcode
site 3 is within the piggyBac TR sequence, immediately adjacent to the TR-Genome junction. Underlined
nucleotides in the 13-bp terminal inverted repeat region (‘CTA’, gold) were targeted for mutagenesis by
mutagenic PCR. (C) Overview of calling card rapid mutagenesis scheme. Mutant amplicons were transfected into
cells with piggyBac transposase and integrated calling cards were collected. Nucleotide frequency for each
mutagenized position of integrated SRTs were calculated. Nucleotide frequency at (D) position 1, (E) position 2
and (F) position 3 of integrated mutated SRTs. Wild-type sequences are outlined in red. All four possible
nucleotides were well-represented at all three mutated positions. IR: internal repeat. TR: terminal repeat.
EF1a: eukaryotic translation elongation factor 1 α promoter. SRT: self-reporting transposon. nt: nucleotide. kb:
kilobase. PuroR: puromycin resistance cassette. WT: wild-type. Mut: mutant.
Figure and caption from:
Matthew Lalli, Allen Yen, Urvashi Thopte, Fengping Dong, Arnav Moudgil, Xuhua Chen, Jeffrey Milbrandt, Joseph D Dougherty, Robi D Mitra, Measuring transcription factor binding and gene expression using barcoded self-reporting transposon calling cards and transcriptomes
The Calling Cards Processing Pipeline
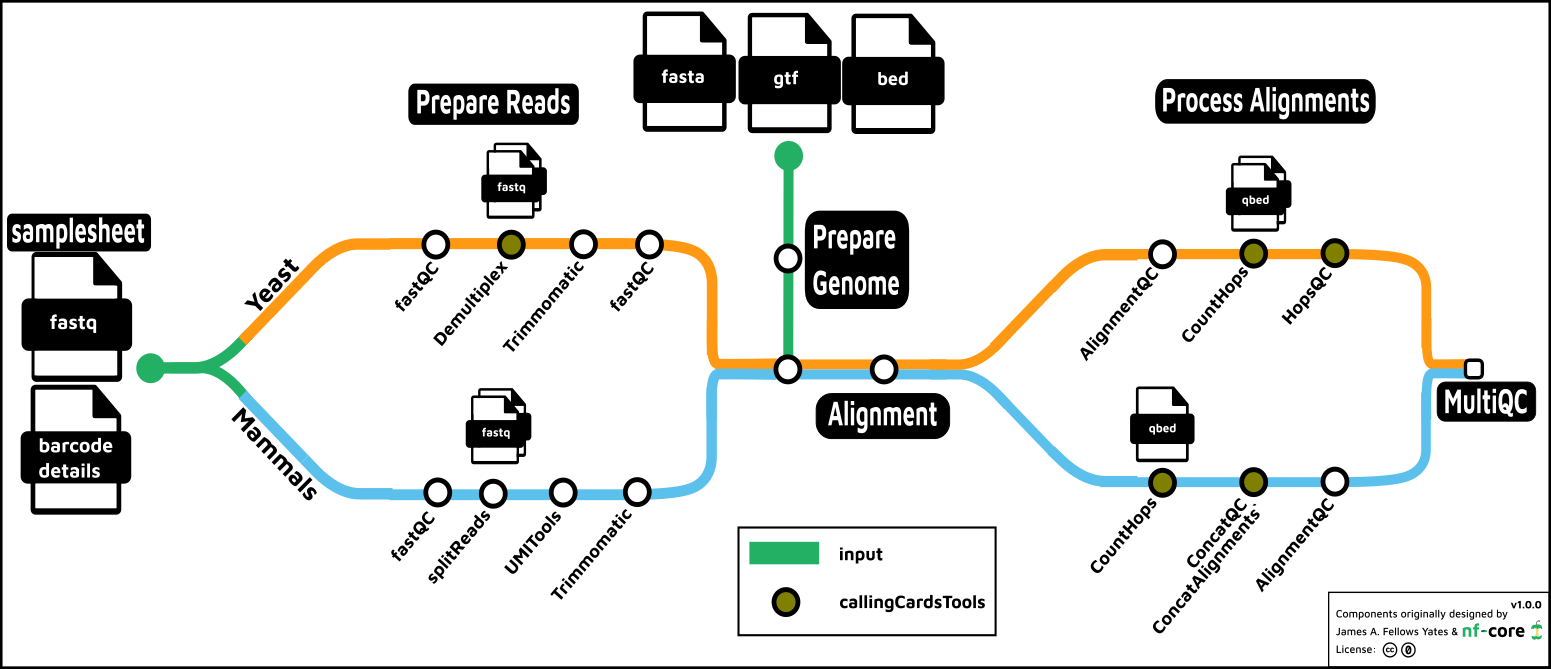
The nf-core/callingcards processing pipeline for yeast
(s. cerevisiae) and mammalian experiments is described in Calling Cards: A Customizable
Platform to Longitudinally Record Protein-DNA Interactions Over Time in Cells and
Tissues by Yen et al.
Briefly, in both the yeast and mammalian systems, the first step is to process reads by trimming off the calling cards barcode sequence. This is appended to the read ID of the fastq file and is utilized in downstream processing. In yeast, if the experiment is multiplexed, the reads are split into separate fastq files according to the barcode which identifies the TF. In both yeast and mammals, the fastq files may be split for parallel processing at this point.
In the next step, the genome is processed according to the selected aligner
(bwa, bwa-mem, bowtie and bowtie2 are the current options). A BED file
describing regions of the genome to hard mask may be included here – this is important
for yeast, in particular, in order to avoid aligning Sir4 reads to the genome
erroneously. However, this BED file is already provided in the pipeline
assets – simply set the -profile yeast in order to correctly set the genome
fasta, gtf and masking regions.
Aftering aligning the reads, the alignments are scanned to validate the calling cards insertions. Preliminary QC metrics are computed, and the data is output in a qBED format, a modified BED6 format, for downstream processing.
Current Development and Future Directions
Current development in the yeast system focuses on the callingCardsTools python package, which handles the calling cards specific read parsing and quantification. More robust QC metrics are in the works, as are recommended methods for handing replicates. The Mitra lab is actively working on the mammalian system, including in single cell based assays.